
Translate this page into:
Recounting COVID-19 associated complement-mediated coagulopathies: Triggers and controls
 , Sadhana Sharma1,
, Sadhana Sharma1,
* Corresponding author: Prof. Sadhana Sharma, Department of Biochemistry, All India Institute of Medical Sciences Patna, Bihar, India. drsadhanas@aiimspatna.org
-
Received: ,
Accepted: ,
How to cite this article: Lata P, Madhuri M, Ali A, Kumari B, Tiwari M, Kumar A, et al. Recounting COVID-19 associated complement-mediated coagulopathies: Triggers and controls. Ann Natl Acad Med Sci (India) 2024;60:244-9. doi: 10.25259/ANAMS_77_2024
Abstract
The coronavirus disease 2019 (COVID-19) pandemic caused by the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) led to a global health crisis, prompting extensive research into its pathogenesis and potential therapeutic interventions. One area of increasing interest is the interaction between SARS-CoV-2 and the complement system, a crucial component of innate immunity. This review explores the intricate relationship between COVID-19 and the complement system, shedding light on how the virus exploits and manipulates the complement components to induce inflammatory responses leading to coagulopathies. The activation of the complement pathway simultaneously activates the coagulation cascade due to the presence of common substrates of mannan-binding serine proteases of the lectin complement system in the coagulation pathway. This cross-talk between the components of the complement and coagulation system further aggravates the dysregulation of immune responses, contributing to the cytokine storm observed in severe COVID-19 cases. A comprehensive understanding of this crosstalk is crucial for developing targeted therapeutic strategies to mitigate the hyperinflammatory state associated with severe disease. Understandings into the molecular mechanisms governing this interaction may pave the way for the development of novel antiviral diagnostics and therapies based on immunomodulatory interventions, offering better management for such cases.
Keywords
COVID-19
SARS-CoV-2
Complement system
Innate immunity
Coagulopathy
Cytokine storm
Therapeutic strategies

INTRODUCTION
In December 2019, Coronavirus disease 19 (COVID-19) originated in Wuhan city in China. The causative agent was severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). It caused 6,945,714 deaths globally, as reported by the World Health Organization (WHO) on June 21, 2023.
COVID-19 was primarily a respiratory illness that affected the lungs and appeared with predominant clinical symptoms of mild fever, cough, and shortness of breath.1 In some patients, it even progressed to life-threatening pneumonia, acute respiratory distress, and multiple organ failure.2
In COVID-19 infection, dysregulated coagulation is a quite common feature which leads to thrombosis in the respiratory, cardiovascular, and venous systems.3 The altered coagulation pattern in COVID-19 patients was termed as COVID-19-induced coagulopathy (CIC), which caused acute cardiovascular complications, potentially leading to death in some patients.4
Different presentations of the coagulopathies in COVID infection
The different forms of thrombotic abnormalities reported in COVID-19 are listed in Table 1. Among these, disseminated intravascular coagulation (DIC)5 and venous thromboembolism (VTE) were the most common,6 which could potentially give rise to strokes and other heart complications.7
| COVID-Associated Thrombotic Complications | Clinical Manifestation |
|---|---|
| Disseminated Intravascular Coagulation (DIC) | Upregulated pro-inflammatory cytokines and plasminogen activator inhibitor-1 (PAI-1), decreased fibrinolysis |
| Venous Thromboembolism (VTE) | Immunological activation of thrombin and hemostasis impairment |
| Thrombotic Microangiopathy (TMA) | CD4 aggregation around thrombotic blood vessels, hemorrhage intense endothelial inflammation |
| Sepsis-Induced Coagulopathy (SIC) | Hypercoagulability, endothelial dysfunction, microthrombosis, and stroke |
DIC is marked by dysregulated systemic activation of the coagulation pathway. The excessive thrombotic and hemorrhagic complications due to intravascular fibrin formation and microangiopathic thrombosis lead to exhaustion of coagulation factors and platelets, resulting in life-threatening hemorrhage.8 VTE is a condition where clot formation occurs inside the veins. In pulmonary embolism, clots in veins break and reach the lungs through bloodstreams. Thrombotic microangiopathy (TMA) is a syndrome comprising a triad of microangiopathic hemolytic anemia, thrombocytopenia, and organ damage. Thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS) are two pro-types of thrombotic thrombocytopenia purpura (TMA). TTP is a life-threatening condition associated with the formation of blood clots in small blood vessels, thus limiting the flow of blood to vital organs such as heart, brain, and kidney. When clot formation occurs in the damaged and inflamed small vessels of the kidney and blocks the filtering system of the kidney, the condition is referred to as HUS; sepsis-induced coagulopathy (SIC) spans from precise activation of coagulation (shown by sensitive markers of the coagulation factor activation) to much sturdy activation of coagulation accompanied by a decrease in platelet count and an increase in clotting time to DIC.9
Pathophysiology of thrombotic events in COVID-19 patients
Angiotensin-converting enzyme 2 (ACE-2) receptors present in different organs (such as lung, kidney, and heart) facilitate entry of SARS-CoV-2 into host cells.10 This phenomenon led to viremia, causing internal injury of the blood vessels and in some cases, ultimately gave rise to severe fatal outcomes such as vascular blockage, thrombosis, and multiple organ failure.11 Entry of the pathogen also induced an inflammatory immune response (cytokine storm) as a preventive measure, which expedited the clotting cascade and negatively resulted in the obstruction of blood vessels.11,12 Upregulation of plasmin inhibitors (alpha-2 antiplasmin and plasminogen activator inhibitor-1) eventually decreased plasmin-mediated fibrinolysis, which resulted in persistent clot accumulation. Further, downregulation of thrombomodulin also induced hypercoagulopathy in COVID-19 patients.13 SARS-CoV-2 viruses use extracellular vesicles for their active migration to nearby or distant sites in a maintained physiological state,14 which are potent inducers of the coagulation system.15,16 von Meijenefeldt et al. (2021) showed a link between COVID-19 and the coagulation pathway, where patients harbor elevated thrombin generation potential and diminished fibrinolytic ability.17 A probable explanation for the hypercoagulable and hypofibrinolytic states was the prolonged activation of the endothelium, which resulted in increased plasma levels of factor VIII and PAI-1.17 Markers of thrombotic events in COVID-associated coagulopathy (CAC) are summarized in Figure 1
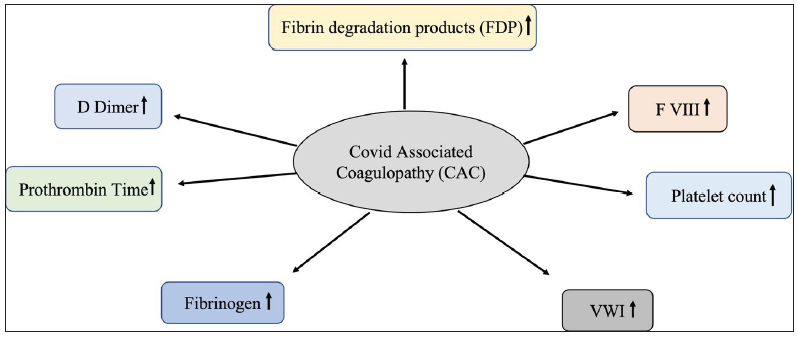
- Markers of coagulopathy during COVID-19 infection: An increase is observed in the levels of fibrin degradation products (FDP), factor VIII (F VIII), platelet count, von Willebrand factor (VWF), fibrinogen, prothrombin time, and D-dimer during SARS-CoV-2 infection.
Aggravated thrombosis in COVID-19 infection, as demonstrated by a couple of studies, suggested the role of autoantibodies in COVID-19. The literature suggests that the antiphospholipid autoantibodies (APL) are also an important factor for the stimulation of endothelial cells and platelets,3,18 although the exact role of these autoantibodies remains unclear.
Upon infection, the viral nucleocapsid and spike proteins trigger the mannose binding lectin (MBL)/Ficolins pathway, which eventually activate lectin pathway proteases such as mannose binding lectin (MBL)-associated serine proteases (MASP)-2, a key factor in inducing prothrombin-mediated clotting.19 COVID-19 patients showed four dysregulated coagulation factors, namely prothrombin (F2), FXI, FXII, and FXIIIa.20 During infection, there is an increase in the level of FXIIIa and von Willebrand factor (VWF) glycoprotein in the renal cortex, which is associated with blood clotting in the renal cortex.21 Serpin Family D Member 1 (SERPIND1) and Serpin peptidase inhibitor (SERPINE1) were downregulated and upregulated, respectively, in the renal cortex, which caused microthrombi formation in COVID-19 patients.20
Significantly higher levels of D-dimer22 and fibrinogen, along with mild thrombocytopenia, are biomarkers of COVID-19-associated hypercoagulopathy23 due to varying underlying reasons.
Crosstalk between complement and coagulation system in COVID-19-associated coagulopathies?
The possible mechanisms involved in coagulopathies with known facts about SARS-CoV-2, the causative agent of contagious COVID-19, are shown in Figure 2. Its spike (S) protein cleavage by furin protease and further priming of the activated S protein by transmembrane protease serine 2 (TMPRSS2) facilitated viral attachment with the host’s ACE-2, which acts as a receptor to mediate its entry into the host cells.24 Therefore, the receptor protein ACE-2 was instrumental in viral transmission and pathogenesis. SARS-CoV-2 interaction with ACE-2 also hinders its physiological role discussed in Figure 2.
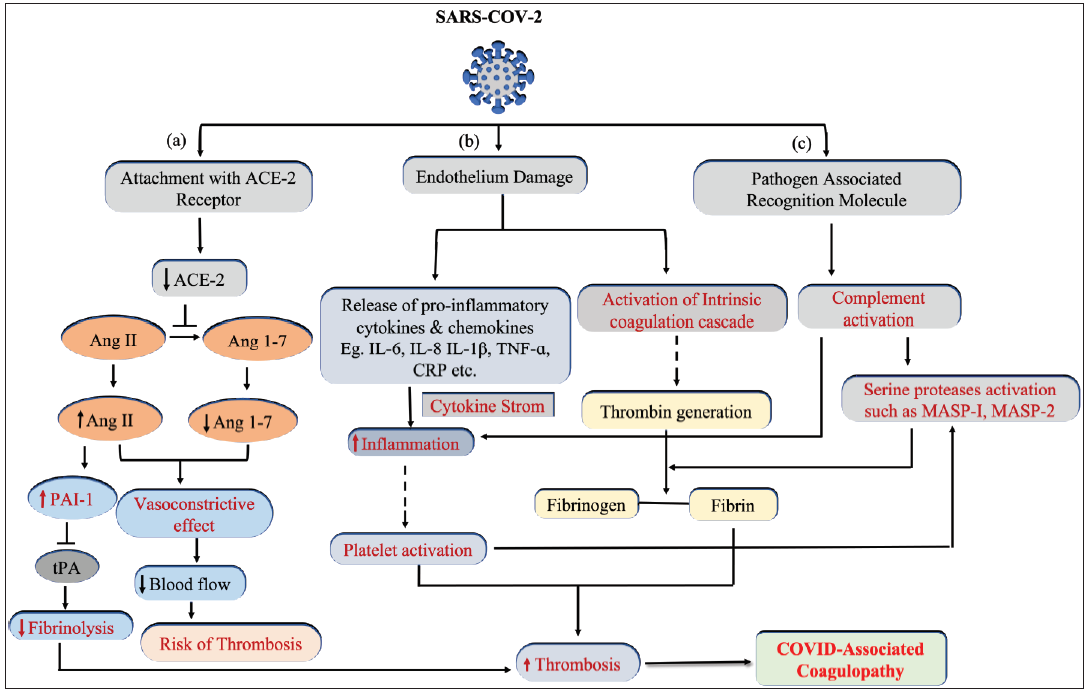
- Schematic representation showing the pathophysiology of COVID-associated coagulopathy. SARS-CoV-2 activates three different pathways. (a) The antithrombotic physiological role of ACE-2 is impaired due to interaction with SARS-CoV-2, resulting in hypofibrinolysis and vasoconstriction. (b) Virus-induced endothelium damage led to simultaneous activation of the classical coagulation cascade and immunothrombosis. (c) Complement activation in response to the recognition of pathogen-associated molecular patterns (PAMPs) contributes to inflammatory thrombosis and nonclassical (MASP protease mediated) fibrin clot generation. ACE: angiotensin-converting enzyme 2, Ang: Angiotensin, MASP: MBL-associated serine proteases, tPA: tissue plasminogen activator, TNF: tumor necrosis factor, CRP: C-reactive protein, IL: interleukin, PAI-1: Plasminogen activator inhibitor-1.
The complement system plays a critical role in augmenting the coagulation process during COVID infection. Activation of proteins of the complement system and coagulation pathway, namely thrombin, trypsin, and plasmin, significantly contributes to COVID-associated thrombotic events.25,26 Previously, an in vivo study with C3 null mice indicated the involvement of the complement pathway in COVID-19 infection.27 Bhagwat et al. (2022) demonstrated that mannan-binding lectin-associated serine proteases (MASP-1 and MASP-2) of the lectin complement pathway also have substrates in the coagulation pathway.28,29 Importantly MASP-1, which has thrombin-like activity, may have played a key role in COVID-19-associated coagulopathies.30 Furthermore, the lectin pathway was also found to be activated in SARS-CoV-2 infection, particularly through the recognition of the nucleocapsid and spike proteins (S protein) of the virus.31 It induced lectin pathway via downstream activation of MASP-1 and 2 leading to thrombotic events.31 Gao et al. (2020) also suggested that in response to SARS-CoV-2, autoactivation of MASP-2 occurred, which eventually activated the lectin pathway via a series of enzymatic cascades.32 In addition, hypercoagulation in COVID-19, marked by VWF,33 was observed consistently, and the literature suggested that it could be the outcome of crosstalk between the complement and coagulation systems.34,35
Furthermore, S protein of SARS-CoV-2 played a key role by binding with heparan sulfate and factor H, which dysregulated the alternative pathway of the complement system.36,37 Moreover, antibodies such as immunoglobin G (IgG) and immunoglobin M (IgM) produced against specific domains of viral spike proteins were capable of activating the classical complement pathway.38 Therefore, SARS-CoV-2 potentially dysregulated or hyperactivated the different components of the complement system, which, in turn, led to endothethilopathy and thromboinflammation, the classical pathophysiology associated with COVID-19.37
Although the exact reasons why other people were predisposed to severe illness while some remained asymptomatic remain undeciphered; the hypercoagulable state and COVID-19 are interconnected [Figure 2].
Complications of complement-mediated coagulopathies in COVID-19: Different forms of thrombotic complications have been reported in clinical patients, which originally initiated in the lungs39 and eventually led to multiple organ failure.40 Pulmonary arterial and venous thrombosis, microangiopathy, and pulmonary embolism were the major clotting-associated complications in COVID-19 patients due to the altered complement system.41,42 Furthermore, diffuse microvascular thrombosis in the capillaries of different vital organs, such as the lung, kidney, and myocardium, along with deposition of complement activation products, were also reported in patients with COVID-19 infection.40,43,44 Niederreiter et al. (2022) showed the deposits of complement proteins, namely C1q, MASP-2, complement factor D, C3c, C3d, and C5b-9, in lungs and kidney samples of COVID-19 patients using immunohistochemical studies, which clearly explains the involvement of the lectin pathway in COVID-19.45 Another study by Carvelli et al. (2020) reported that MASP-2, C4, MAC, and macrophages with overexpressed C5aR1 are associated with endothelium damage and microthrombi formation.46 Clinical studies with critically ill COVID-19 patients demonstrated significantly elevated levels of circulating markers of complement activation. In addition, upregulated alternative pathways along with markers of endothelial injury (i.e., angiopoietin-2) and hypercoagulability (i.e., thrombomodulin and VWF) were reported as the most common features of COVID-19 patients.33
Systemic activation of the complement and coagulation cascade in response to the viral pathogen also promotes localized complement protein production by lung cells.37 Therefore, complement-targeting therapies have been developed as an attractive therapeutic strategy for COVID-19. Although the pernicious effects of hyperactivated complement pathways in COVID-19 are quite evident, an in-depth understanding of the driving molecular mechanisms is not there. Several studies have demonstrated that the complement system, especially the lectin pathway, is indeed instrumental in viral illness and plays a crucial role in COVID-19 pathogenesis.
CONCLUSION
The COVID-19 pandemic has emerged as a challenging threat to global health in this century. A comprehensive understanding of disease pathophysiology, risk factors, and therapeutic regimens is essential for disease management in future episodes. This knowledge will have beneficial effects in future research for disease control and prevention.
However, the molecular mechanisms underlying the pathogenesis still require attention. Extensive multiomics approach may help us in obtaining an in-depth understanding and may provide new insights for the development of novel prophylactic and therapeutic strategies to combat this viral illness.
Authors’ contributions
PL, MM and AA: conceptualization, writing the original draft, BK and MT: writing-review, and editing, AK: coordinated revisions, KH and DB: conceptualization, writing-review, and editing. SS: conceptualization, writing the original draft, writing-review, and final editing. All authors approved the submitted version.
Ethical approval
Institutional Review Board approval is not required.
Declaration of patient consent
Patient’s consent not required as there are no patients in this study.
Financial support and sponsorship
AIIMS Patna, Intramural grant Ref. No. RC/AIIMS/Pat/2020/528
Conflicts of interest
There are no conflicts of interest.
Use of artificial intelligence (AI)-assisted technology for manuscript preparation
The authors confirm that there was no use of artificial intelligence (AI)-assisted technology for assisting in the writing or editing of the manuscript and no images were manipulated using AI.
REFERENCES
- Coronavirus disease (COVID-19): Comprehensive review of clinical presentation. Front Public Health. 2021;8:582932.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- A Review of coronavirus disease-2019 (COVID-19) Indian J Pediatr. 2020;87:281-6.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Coagulation disorders and thrombosis in COVID-19 patients and a possible mechanism involving endothelial cells: A review. Aging Dis. 2022;13:144-56.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- COVID-19 induced coagulopathy (CIC): Thrombotic manifestations of viral infection. TH Open. 2022;6:e70-e79.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- COVID-19-associated coagulopathy and disseminated intravascular coagulation. Int J Hematol. 2021;113:45-57.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Thromboembolism risk of COVID-19 is high and associated with a higher risk of mortality: A systematic review and meta-analysis. EClinicalMedicine. 2020;29:100639.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Laboratory diagnosis of COVID-19: current issues and challenges. J Clin Microbiol. 2020;58:e00512-20.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Sepsis-induced coagulopathy: An update on pathophysiology, biomarkers, and current guidelines. Life (Basel). 2023;13:350.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Coagulopathy and sepsis: Pathophysiology, clinical manifestations and treatment. Blood Rev. 2021;50:100864.
- [CrossRef] [PubMed] [Google Scholar]
- New insights on the antiviral effects of chloroquine against coronavirus: What to expect for COVID-19? Int J Antimicrob Agents. 2020;55:105938.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- COVID-19 and multiorgan failure: A narrative review on potential mechanisms. J Mol Histol. 2020;51:613-28.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The nature and extent of COVID-19 vaccination hesitancy in healthcare workers. J Community Health. 2021;46:1244-51.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Persistent clotting protein pathology in Long COVID/Post-Acute Sequelae of COVID-19 (PASC) is accompanied by increased levels of antiplasmin. Cardiovasc Diabetol. 2021;20:172.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Circulating exosomes are strongly involved in SARS-CoV-2 infection. Front Mol Biosci. 2021;8:632290.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Patients with COVID-19 have elevated levels of circulating extracellular vesicle tissue factor activity that is associated with severity and mortality-brief report. Arterioscler Thromb Vasc Biol. 2021;41:878-82.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Dissemination of extreme levels of extracellular vesicles: Tissue factor activity in patients with severe COVID-19. Blood Adv. 2021;5:628-34.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Sustained prothrombotic changes in COVID-19 patients 4 months after hospital discharge. Blood Adv. 2021;5:756-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Antimicrobial resistance and COVID-19: Intersections and implications. Elife. 2021;10:e64139.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Role of classical complement activation in SARS-Cov-2 infection; A case report of COVID-19 in a patient with saddle pulmonary embolism. J Medical Case Reports Case Series 2020:1-3. Available from: https://www.acquaintpublications.com/article/role_of_classical_complement_activation_in_sars_cov_2_infection_a_case_report_of_covid_19_in_a_patient_with_saddle_pulmonary_embolism83 [Last accessed 18 April 2024]
- [Google Scholar]
- Multi-organ proteomic landscape of COVID-19 autopsies. Cell. 2021;184:775-791.e14.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Role of von Willebrand factor in the haemostasis. Blood Transfus. 2011;9 Suppl 2:s3-8.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Molecular architecture of the SARS-CoV-2 virus. Cell. 2020;183:730-738.e13.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- COVID-19, immunothrombosis and venous thromboembolism: Biological mechanisms. Thorax. 2021;76:412-20.
- [CrossRef] [PubMed] [Google Scholar]
- SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271-280.e8.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Anti-C5a antibody IFX-1 (vilobelimab) treatment versus best supportive care for patients with severe COVID-19 (PANAMO): An exploratory, open-label, phase 2 randomised controlled trial. Lancet Rheumatol. 2020;2:e764-e773.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Generation of C5a in the absence of C3: A new complement activation pathway. Nat Med. 2006;12:682-7.
- [CrossRef] [PubMed] [Google Scholar]
- Complement activation contributes to severe acute respiratory syndrome coronavirus pathogenesis. mBio. 2018;9:e01753-18.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Fibrinogen cleavage by lectin pathway proteases: Roles and implications. Available from: https://www.austinpublishinggroup.com/ [Last accessed 18 April 2024]
- [Google Scholar]
- Identification of substrates of MBL Associated Serine Protease-1 (MASP-1) from human plasma using N-terminomics strategy. Mol Immunol. 2022;151:114-25.
- [CrossRef] [PubMed] [Google Scholar]
- COVID-19-related coagulopathy: A review of pathophysiology and pharmaceutical management. Cell Biol Int. 2021;45:1832-50.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Lectin pathway mediates complement activation by SARS-CoV-2 proteins. Front Immunol. 2021;12:714511.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Highly pathogenic coronavirus N protein aggravates lung injury by MASP-2-mediated complement over-activation. MedRxiv 2020 Mar 30:2020-03
- [Google Scholar]
- Global percentage of asymptomatic SARS-CoV-2 infections among the tested population and individuals with confirmed COVID-19 diagnosis: A systematic review and meta-analysis. JAMA Netw Open. 2021;4:e2137257.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Hyperinflammatory shock in children during COVID-19 pandemic. Lancet. 2020;395:1607-8.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- An outbreak of severe Kawasaki-like disease at the Italian epicentre of the SARS-CoV-2 epidemic: An observational cohort study. Lancet. 2020;395:1771-8.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Direct activation of the alternative complement pathway by SARS-CoV-2 spike proteins is blocked by factor D inhibition. Blood. 2020;136:2080-9.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- The state of complement in COVID-19. Nat Rev Immunol. 2022;22:77-84.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Systemic complement activation is associated with respiratory failure in COVID-19 hospitalized patients. Proc Natl Acad Sci U S A. 2020;117:25018-25.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- COVID-19 and coagulation: Bleeding and thrombotic manifestations of SARS-CoV-2 infection. Blood. 2020;136:489-500.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Microthrombi as a major cause of cardiac injury in COVID-19: A Pathologic Study. Circulation. 2021;143:1031-42.
- [CrossRef] [PubMed] [Google Scholar]
- Mechanisms of COVID-19 associated pulmonary thrombosis: A narrative review. Biomedicines. 2023;11:929.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Pulmonary venous thrombosis in a patient with COVID-19 infection. J Thromb Thrombolysis. 2021;51:985-8.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Pulmonary vascular endothelialitis, thrombosis, and angiogenesis in Covid-19. N Engl J Med. 2020;383:120-8.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Complement activation in kidneys of patients with COVID-19. Front Immunol. 2021;11:594849.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Complement activation via the lectin and alternative pathway in patients with severe COVID-19. Front Immunol. 2022;13:835156.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]
- Association of COVID-19 inflammation with activation of the C5a-C5aR1 axis. Nature. 2020;588:146-50.
- [CrossRef] [PubMed] [PubMed Central] [Google Scholar]



