
Translate this page into:
Cystatin A Down-regulation in Head and Neck Squamous Cell Carcinoma Cell Lines Decreases Cancer Hallmark Signatures

Address for correspondence Dr. Arnab Pal, MD, PhD, Department of Biochemistry, Post Graduate Institute of Medical Education and Research, Chandigarh 160012, India (e-mail: pal.arnab@pgimer.edu.in).
This article was originally published by Thieme Medical and Scientific Publishers Pvt. Ltd. and was migrated to Scientific Scholar after the change of Publisher.
Abstract
Background
Cystatin A (CSTA), an endogenous inhibitor of lysosomal cysteine protease, is expressed primarily in epithelial tissues. The expression of CSTA was found to be dysregulated in various cancers and associated with cancer pathogenesis, but its role is reported to be contradictory. Our previous preliminary study found CSTA to be upregulated in the saliva and tissues of patients with head and neck squamous cell carcinoma (HNSCC). In this current study, we have explored the role of CSTA in the pathophysiology of HNSCC.
Methods
First, we confirmed the upregulation of CSTA in CAL 27 (p = 0.0242) and FaDu (p = 0.0014), two HNSCC cell lines, compared to the normal gingival epithelium. CSTA was then stably knocked down in CAL 27 and FaDu using the lentiviral short hairpin RNA pLKO vector transduction to study the effects of CSTA knockdown on various cancer hallmarks such as cell proliferation ability, invasion, migration, colony formation, and chemotherapy-induced apoptosis.
Results
CSTA knockdown significantly decreased cell viability, cell migration, transwell invasion, and colony formation in both cell lines. CSTA downregulation also enhanced cisplatin-induced apoptosis.
Conclusion
Overall, this study suggests the protumorigenic role of CSTA in HNSCC.
Keywords
cystatin A
HNSCC
shRNA pLKO vector transduction
cancer hallmarks

Introduction
Head and neck squamous cell carcinoma (HNSCC) arises from the buccal cavity and upper aerodigestive tract carcinoma and is one of the most common cancers worldwide1. There have been significant advancements in diagnostic and treatment strategies for HNSCC in recent years, even though the 5-year survival rate remained abysmal.2 This suggests that a better understanding of cancer cells' biological and molecular characteristics must be studied to design newer prevention and therapeutic strategies for HNSCC. Therefore, there is an urgent need to relook into the biological pathways/proteins dysregulated in a specific way that may cause the tumor progression in HNSCC.
The perturbation of various proteins due to the cancer cell's protective mechanisms or oncogenic changes in the genome and/or epigenome of the cancer cell is a known phenomenon in oncogenesis.2 In both cases, detecting and quantifying specific proteins provide important information regarding the origin of oncogenic changes, disease staging, progression, and patient outcome. In our previous findings, various proteins in the saliva of HNSCC patients were found to be dysregulated using liquid chromatography-mass spectrometry analysis. We identified that levels of cystatin A (CSTA) were significantly raised in the saliva of HNSCC patients compared to controls (data not shown). We also found high cytoplasmic expression of CSTA in the tumor cells of HNSCC patients (►Supplementary Material Fig. 1, available in the online version).
In the literature, the contradictory role of CSTA has been reported in different normal tissues and cancers. CSTA belongs to type I cystatins, is a cytoplasmic inhibitor of cathepsins B, H, and L (cysteine proteases), and is primarily expressed in epithelial and lymphoid tissues.3 CSTA is a cytoplasmic protein found in body fluids when upregulated.4 In nasopharyngeal carcinoma, a high level of CSTA in patients' serum is related to poor prognosis, whereas prostate cancer with high CSTA expected to be less aggressive.5,6 High extracellular CSTA levels in colorectal carcinoma were related to short patient survival, indicating CSTA's role in tumor progression.7,8 There is a contradiction in the role of CSTA in HNSCC pathophysiology. In some studies, the level of CSTA was found to be downregulated, while in the human protein atlas, an enhanced level of CSTA has been reported in HNSCC.9 In oesophagal squamous cell carcinoma, decreased levels of CSTA in tumor mucosa are associated with nodal involvement and overall bad prognosis.10,11
A few studies have explored CSTA's role in HNSCC pathophysiology. We hypothesized that CSTA might play a crucial role in HNSCC pathophysiology. Modulating CSTA in the HNSCC cell line enabled us to study its effect on cancer hallmarks like cell proliferation, apoptosis, migration, and invasion and generate preliminary information on its role in HNSCC pathophysiology.
Materials and Methods
Cell Lines
STR-matched cell lines—FaDu (ATCC HTB-43) and CAL 27 (ATCC CRL—2095) were used in this study. FaDu is a cell line of pharyngeal squamous cell carcinoma, while CAL 27 is a cell line of tongue squamous cell carcinoma. HEK 293T (ATCCCRL-3216) cell line was used to generate lentiviral particles.
Maintenance of Cell Line
The cell lines were grown in Dulbecco's Modified Eagle Medium (DMEM) media (Lonza,12-604F) containing 10% fetal bovine serum (FBS) and 1X Antibiotic-Antimycotic (Gibco,15-240-062) in a humidified incubator with 5% CO2 at 37°C. Passaging of cells was done once they reached approximately 60 to 80% confluency. The cell monolayer was given a wash with phosphate buffered saline (pH = 7.4) to take off the residual serum. Then, adherent cells were harvested with trypsin (0.25%) (Gibco, 25200056). The media was changed every 3rd day. All culture work was done strictly under aseptic conditions in the tissue culture hood with vertical laminar flow.
mRNA Expression Studies
For mRNA expression studies, total RNA was isolated using QIAzol Lysis Reagent (Qiagen,79306) and followed by synthesis of cDNA using a cDNA synthesis kit by Thermo Scientific (AB1453B). Quantitative polymerase chain reaction (qPCR) for CSTA was done using SYBR green chemistry in Bio-Rad CFX96 Real-Time qPCR System with 18S rRNA as reference gene. The details of the primers and the amplification curves are given in ►Supplementary Material Table 1 and ►Supplementary Material Fig. 2 (available in the online version), respectively.
CSTA Short Hairpin RNA Expression Vector Construction and Stable Transduction by Lentivirus
The CSTA gene was knockdown by stably expressing a short hairpin RNA (shRNA) against the CSTA mRNA, using lentiviral transduction. A 3rd-generation lentiviral negative control vector containing scrambled shRNA from add gene was used as a control vector. shRNA oligos against CSTA cloned in pLKO.1 vector was taken from the shRNA library of BROAD INSTITUTE (https://portals.broadinstitute.org/gpp/public/dir?dirpath=shrna_annot/legacy) (►Supplementary Material Table 2, available in the online version), and scrambled shRNA was taken from David Sabatini (Addgene plasmid # 184; http://n2t.net/addgene:1864; RRID: Addgene_1864) (►Supplementary Material Table 3, available in the online version). Both of the plasmids were already transformed into One Shot TOP10 Competent E. coli cells (C4040-10). These E. coli cells were cultured, and the plasmid DNA was isolated using Promega PureYield Plasmid Miniprep System (A1223).
Generation of Lentiviral Particles
HEK-293T was cultured in 6-well plates for each plasmid to be transfected using Lipofectamine 3000 Transfection Reagent (L3000001). The cells were regularly passaged in DMEM with 10% FBS and 1X Antibiotic-Antimycotic.
For transfection, a mix was prepared using the following for each plasmid:
shRNA plasmid = 500 ng (pLKO.1)
Packaging plasmid = 375 ng (psPAX2)
Envelope plasmid = 125 ng (pMD2.G)
3 μL of P3000 Reagent
125 μL serum-free OPTI-MEM
This mix was then added to the diluted Lipofectamine 3000 Reagent (following the manufacturer's protocol) and then incubated at room temperature for 30 minutes. This mix was added to 60 to 70% confluent HEK 293T cells cultured in DMEM with 5% FBS. After 12 to 16 hours, the media was replaced by complete DMEM with 10% FBS and 1X Antibiotic-Antimycotic—3 mL each well. The viral titers were collected at 24 and 36 hours and stored at −80 degrees until further use. These lentiviral particles were used for the transduction of cell lines.
Transduction of Cell Lines
The cell lines, FaDu and CAL 27, were cultured in 6-well plates using complete DMEM with 10% FBS and 1X Gibco Antibiotic-Antimycotic. Transduction was done at 70% confluency of cells. The lentiviral titer was diluted with DMEM media (1:1) without any antibiotic and had 8 μg/mL polybrene. This mixture (2 mL) was added to each one of the wells and then incubated for 24 hours. After 24 hours, a fresh complete DMEM with 10 μg/mL puromycin was added as the selected antibiotic. These cells were then continuously grown in the puromycin-containing media (10 μg/mL). First, we used four CSTA shRNA clones (namely C7, C9, C10, and C11) for transduction, but we proceeded with the C11 clone as this showed maximum CSTA gene knockdown analyzed by mRNA expression.
Effect of Cystatin A Downregulation on Cancer Hallmarks in Head and Neck Squamous Cell Carcinoma Cell Line(s)
Various assays were performed to analyze cancer hallmarks like proliferation, migration, apoptosis, invasion and anchorage-independent growth in CSTA shRNA expressing vector, scrambled control (mock-treated cell lines), and the cell line having no transduction (parent or untreated cell lines). All of the experiments were repeated at least three times.
Cell Viability Assay
Cell viability was assessed by using MTT assay. The assay was performed 36 hours after seeding the cells in 6-well plates (detailed methodology added in supplementary data, available in the online version).
Cell Death Assay (Annexin V/PI Apoptotic Death Assay)
FITC Annexin V/Dead cell Apoptosis Kit (Invitrogen, V13242) was used. The cells were first treated with an LD50 dose of cisplatin for 24 hours to analyze the effect of CSTA knockdown on cisplatin-induced apoptosis—a widely used anticancer drug. The LD50 for CAL 27 was 7.5 μM and for FaDu was 3.72 μM used.12 After 24 hours of the drug treatment, cells were incubated with annexin V and propidium iodide dye according to the manufacturer's protocol, and tubes were immediately analyzed by flow cytometry.
Cell Migration Assay
Scratch assay was done to analyze the migration of cancer cells. A scratch was made using a 10-μL pipette tip in the form of a cross through the cell monolayer at 80 to 90% confluency. Pictures of the scratch were taken at various time intervals using a bright field microscope. The time point at which the scratch was made, denoted as T0, and pictures were taken after 48 hours of scratch making (T48) (details in supplementary data, available in the online version).
Soft Agar Colony-Forming Assay
To determine the anchorage-independent proliferative capacity of cancer cells, soft agar colony formation was assessed. For this, cells were cultured in the agar of 0.3%, and another agar of 0.5% was layered beneath to cells to prevent adhesion to the culture plate. 5 × 103 cells of each type were counted, seeded in the top agar layer, and incubated for 15 days. The colonies were stained by crystal violet and counted under a 10x resolution bright field light microscope.
Cell Invasion Assay
The assay was carried out using Transwell Corning Matrigel matrix in the Boyden chamber with Matrigel in the upper chamber with low FBS (5%) media and lower chamber with high FBS (10%) media. Cells were counted, and 1 × 103 were resuspended in the upper chamber. After 24 hours of incubation, cells that invaded the transwell were stained using crystal violet and enumerated under a bright field light microscope at 10x resolution.
Statistical analysis: The results were analyzed by using GraphPad Prism V9. The nonparametric version of one-way ANOVA, that is, the Kruskal–Wallis test was used among different groups. p ≤ 0.05 is taken as the level of significance.
Results
CSTA was upregulated in HNSCC cell lines: The gene expression of CSTA in two HNSCC cell lines, CAL 27 and FaDu, was analyzed by real-time quantitative reverse transcription polymerase chain reaction. CSTA was significantly upregulated in both HNSCC cell lines, CAL 27 (p = 0.0248*) and FaDu (p = 0.0015**), in comparison to normal gingival epithelial tissue in ►Fig. 1a.
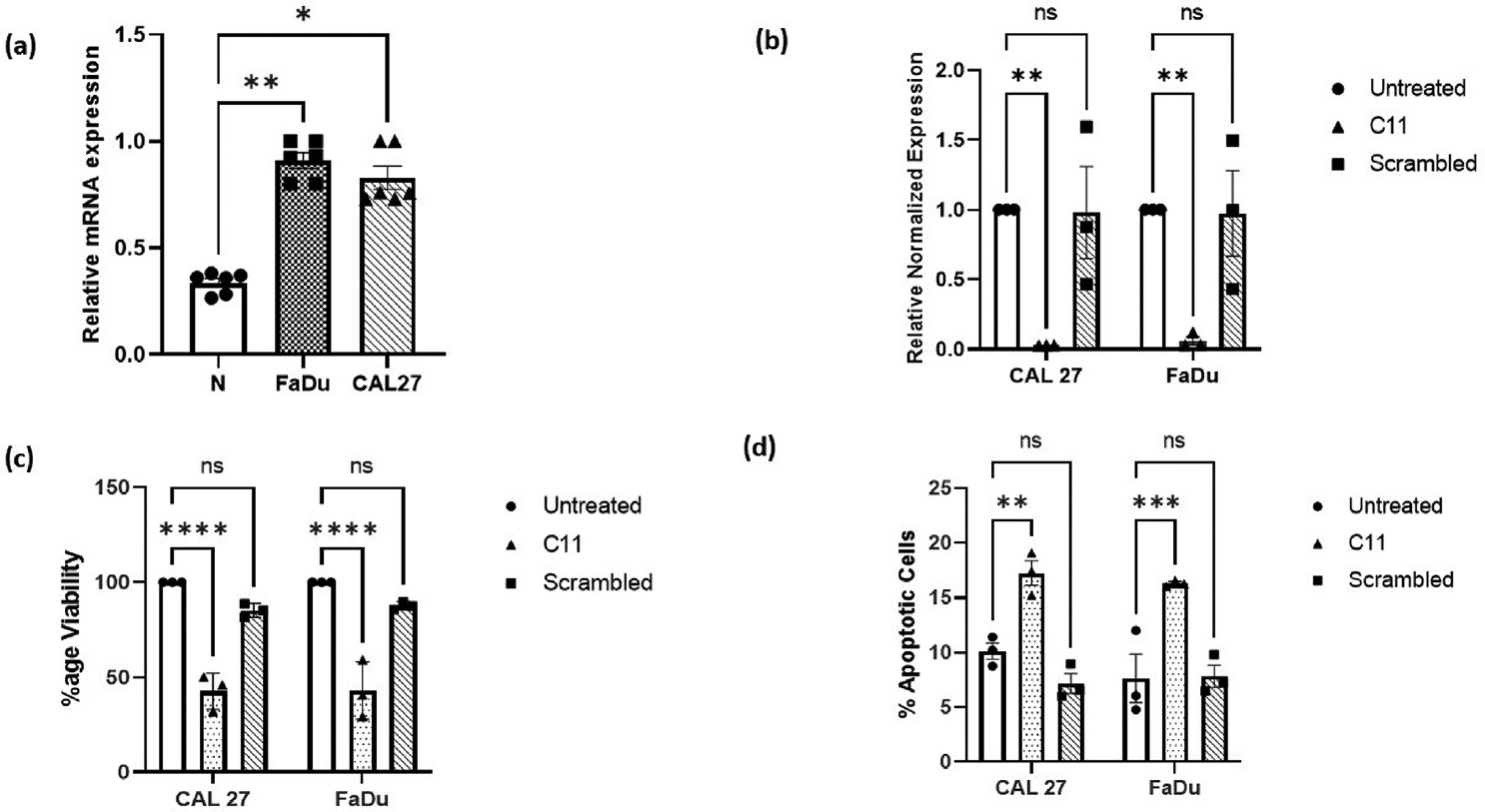
- (a) Cystatin A gene (CSTA) expression was analyzed by qRT-PCR. CSTA mRNA was significantly upregulated in CAL 27 (p = 0.0248*) and in FaDu (p = 0.0015**) cell lines as compared to normal gingival epithelium tissue (N). Fig 1(b) Cystatin A gene expression levels in CAL 27 and FaDu stable cell lines were analyzed by qRT-PCR. CSTA gene was significantly downregulated by C11 CSTA shRNA-expressing vector, both in CAL 27 (p = 0.0054**) and FaDu (p = 0.0067 **) cell lines as compared to untreated cell lines. Fig 1(c) MTT assay revealed that the percentage viability of cells was significantly decreased both in C11 CAL 27 (p <0.0001****) and FaDu (p <0.0001****) in comparison with that of untreated. There was no significant change in the cell viability in case of scrambled cell lines. Fig 1(d) It was observed that the cisplatin-induced apoptosis was significantly increased in the CAL 27 (p = 0.0025**) and FaDu (p = 0.0005***) cell lines upon the knockdown of CSTA as compared to the untreated cell line. The induction of apoptosis in scrambled control was almost similar to that of untreated.
CSTA was stably downregulated in HNSCC cell lines: The two HNSCC cell lines, that is, CAL 27 and FaDu, were transduced by lentiviral particles having CSTA shRNA and scrambled shRNA expressing vector (scrambled act as mock-treated cell lines). Initially, there were four clones of CSTA shRNA expressing vector. Still, CSTA was significantly down-regulated in the C11 clone in both of the cell lines, CAL 27 (p = 0.0054**) and FaDu (p = 0.0067**), in comparison to respected wild-type cell lines (►Supplementary Material Fig. 3, available in the online version). Also, there was no significant change in the gene expression level of CSTA in scrambled shRNA-treated cell lines in ►Fig. 1b. A similar downregulation of CSTA protein expression was also found by immunocytochemistry in the shRNA-treated cells (►Supplementary Material Fig. 4, available in the online version)
CSTA downregulation is associated with a decrease in cell viability: To analyze the knockdown effect of CSTA on cell viability, an MTT assay was performed in the two cell lines. It was found that in the cell lines (both in CAL 27 and FaDu) in which CSTA was stably knockdown, the cell viability was significantly reduced (p < 0.0001****) as compared to untreated and scrambled shRNA-treated cell lines shown in ►Fig. 1c.
CSTA downregulation enhances cisplatin-induced apoptosis: Cisplatin is an important chemotherapeutic agent for treating HNSCC. To analyze the effect of CSTA downregulation on cisplatin-induced apoptosis, a flow cytometry-based annexin V/PI assay was performed. Cells treated with H2O2 were taken as a positive control of apoptosis. In both the cell lines CAL 27 and FaDu, in which CSTA was downregulated, the percentage of apoptotic cells was significantly increased (p = 0.0025** and p = 0.0005***, respectively), as compared to the nontransduced (untreated) and scrambled shRNA-treated cells shown in ►Fig. 1d. This indicates that CSTA downregulation potentiates apoptosis induced by cisplatin.
Downregulation of CSTA is correlated with reduced cancer cell migration: Cell migration is one of the cancer hallmarks. To analyze the effect of CSTA downregulation on the migratory property of cancer cells, a monolayer wound healing/scratch assay was performed. The rate of scratch filling was observed at different time intervals. It was found that cells transduced with CSTA shRNA expressing vector spread much more slowly in comparison to the untreated group in CAL 27 (p = 0.0002***) and FaDu (p < 0.0001****) given in ►Fig. 2a,b.

- (A) In wound healing/scratch assay, a scratch was made across the monolyer and images were take at 0 hour and at 48 hours after the scratch formation in the phase microscope under 10X resolution. (B) In monolayer wound healing/scratch assay, the rate of scratch filling was significantly reduced in CSTA knockdown CAL 27 (p = 0.0002***) and FaDu (p < 0.0001****) cell lines in comparison to the untreated group. (C) The invaded cells in the Boyden Chamber transwell invasion assay were quantified under a brightfield microscope at 10x resolution. (D) The effect of CSTA knockdown on invasion of cancer cells was analyzed by the Boyden Chamber transwell invasion assay. The invaded cells were quantified after 24 hours of seeding by microscope under 10X resolution. The number of invaded cells were significantly reduced upon CSTA gene knockdown in CAL 27 (p = 0.0491*)and FaDu (p = 0.0001***) as compared to the untreated cell lines. (E) Colony-forming assay, the cells were grown in an agar layer and incubated for 15 days. The number of colonies formed were stained by crystal violet and visualized under a bright field microscope. (F) In colony-forming assay, the number of colonies formed by the cells transduced with CSTA shRNA expressing vector was significantly low as compared with the both parent cell lines, Cal 27 (p ≤ 0.0013**) and FaDu (p ≤ 0.0003***).
CSTA knockdown significantly reduced transwell invasion of cancer cells: Boyden Chamber transwell invasion assay was done to analyze the impact of CSTA downregulation on cancer cell invasiveness. In this experiment, the invaded cells were quantified under a microscope, and the results were similar to the cell migration assay. It was found that the number of cells that have invaded was significantly reduced upon CSTA gene knockdown in CAL 27 (p = 0.0491*) and FaDu (p = 0.0001***) in comparison to the wild-type cell lines shown in ►Fig. 2c, d.
CSTA downregulation inhibits colony formation in HNSCC cell lines: To analyze the ability of transduced cells to form colonies in an anchorage-independent manner, colony-forming assay was done. The results revealed that the number of colonies established by the cells transduced with CSTA shRNA expressing vector was significantly low as compared with the parent both in Cal 27 (p ≤ 0.0013**) and FaDu (p ≤ 0.0003***) shown in ►Fig. 2e,f.
Discussion: HNSCC are the most common cancers globally. The 5-year survival rate in HNSCC patients is below 50% despite development in various cancer management strategies.1 Therefore, it becomes crucial to understand the pathophysiology of HNSCC up to the molecular level.
Our group identified various dysregulated proteins in HNSCC patients' saliva using the liquid chromatography with tandem mass spectrometry approach. Among them, CSTA was significantly elevated in the saliva of patients with HNSCC compared to the controls.
CSTA is a cytoplasmic protein and an inhibitor of cysteine proteases, specifically lysosomal cysteine proteases (cathepsin B, H, K, and L). Lysosomal cathepsins promote cancer metastasis and invasion by degrading the extracellular matrix (ECM). CSTA is a tumor suppressor in some cancers, inhibiting some cathepsins' activities and preventing cancer metastasis.9,10,11,13,14,15,16,17,18
Luo et al found that CSTA levels in esophageal squamous cell carcinoma (ESCC) patients decreased in tumor tissue compared to normal tissue and were related to tumor progression and poor prognosis.10 In contrast to the above study, a more recent study by Shiba et al demonstrated that increased levels of CSTA in ESCC were associated with locoregional metastasis and tumor progression with the advanced cancer stage.11 Similar contradictory results were also found in other cancers and HNSCC. Also, higher levels of CSTA were associated with the increased survival probability of HNSCC patients (Human Protein Atlas accessed on 02/03/2023).
Studies have shown that CSTA may play a dual role in the pathogenesis of HNSCC. It can be inferred from the literature that CSTA may play an additional role other than cysteine protease inhibitor.18 However, the exact role of CSTA in the pathophysiology of HNSCC is still not clear.
In our study, CSTA was knocked down in two HNSCC cell lines, and its effect on various cancer hallmarks was elucidated in vitro.
We found significantly higher levels of CSTA in FaDu and CAL 27 cell lines compared to normal healthy oral gingival tissue. CSTA mRNA expression was successfully downregulated by lentiviral transduction of CSTA shRNA in FaDu and CAL 27 cell lines. Interestingly, it was found that cell viability after CSTA downregulation was significantly reduced. One possible mechanism behind this reduced viability may be apoptotic death, so we performed an annexin V/propidium iodide cell death assay. CSTA's knockdown significantly increased cisplatin-induced apoptosis in both HSNCC cell lines, that is, CAL 27 and FaDu. Also, in the literature, a study reported that high expression of CSTA in tumor cells attenuates tumor necrosis factor -induced apoptosis along with cathepsin activity.11 This suggests that an increased expression of CSTA prevents cancer cells from apoptosis. Hence, the downregulation of CSTA may potentiate apoptosis through chemotherapeutic agents like cisplatin.
Cancer cells grow anchorage independent, while normal epithelial cells undergo anoikis following detachment from the surrounding ECM. By performing a soft agar colony formation assay, it was observed that cells with downregulated CSTA showed a reduction in the number of colonies formed in the agar layer, indicating that CSTA downregulation reduces anchorage-independent growth in cancer cells.
Cancer cell migration and metastasis are important steps in cancer progression. The scratch assay and transwell invasion assay were performed to analyze the effect of CSTA downregulation on cancer cells' migration and invasion properties. The results of both assays showed that the property of migration and invasion was significantly reduced after CSTA knockdown, showing that CSTA may be involved in tumor progression as its downregulation reverse the processes.
From our study, it can be suggested that CSTA down-regulation reduces various cancer hallmarks, viz. cell viability, cell proliferation, cell migration, anchorage-independent growth, and significantly increased cell death by apoptosis in the in vitro model.
A study by Shiba et al on ESCC showed that the proliferation marker, Ki67, was highly expressed in the CSTA-expressing cancer cells. In normal mucosa, the Ki67-expressing cells do not express CSTA, suggesting that CSTA up-regulation may be advantageous for cancer cells in terms of an increase in proliferation, thus supporting our results11.
Our results have shown the protumorigenic role of CSTA in HNSCC pathophysiology, suggesting the possibility of CSTA targeting in cancer treatment. However, the results must be further validated in more HNSCC cell lines, primary culture, and in-vivo models. Also, the mechanistic studies for elucidating the pathways affected by CSTA in the progression of HNSCC need to be well explored.
Ethics Approval Statement
This study was performed after getting the approval of the Institute Ethics Committee, PGIMER, Chandigarh.
Data Availability Statement
Detailed data and protocols shall be made available by the corresponding author on reasoned request.
Authors' Contribution
G.S.B., A.C., and A.P. conceived and planned the experiments. G.S.B. carried out the experiments. G.S.B. wrote the manuscript with input from all authors. A.B. analyzed the IHC and ICC slides. G.S.B., A.C., A.B, R.K.V., and A.P. contributed to the analysis and interpretation of the results. A.C. and A.P. edited the manuscript
Acknowledgment
We acknowledge the help of Ms Rajandeep Kaur and Dr Shabir Ahmad Bhat for standardizing the transduction experiments.
Conflict of Interest
None declared.
Funding
This work was supported by PGIMER Special Research Grant for funding the study and Council for Scientific and Industrial Research (CSIR), New Delhi, for providing the fellowship to AC.
References
- Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71(03):209-249.
- [CrossRef] [PubMed] [Google Scholar]
- Clinical significance of head and neck squamous cell cancer biomarkers. Oral Oncol. 2014;50(03):168-177.
- [CrossRef] [PubMed] [Google Scholar]
- The cystatins: a diverse superfamily of cysteine peptidase inhibitors. Biomed Biochim Acta. 1986;45(11-12):1363-1374.
- [Google Scholar]
- Protein inhibitors of cysteine proteinases. I. Isolation and characterization of stefin, a cytosolic protein inhibitor of cysteine proteinases from human polymorphonuclear granulocytes. Hoppe Seylers Z Physiol Chem. 1983;364(11):1475-1480.
- [CrossRef] [PubMed] [Google Scholar]
- Ratio of cathepsin B to stefin A identifies heterogeneity within Gleason histologic scores for human prostate cancer. Prostate. 2001;48(04):274-284.
- [CrossRef] [PubMed] [Google Scholar]
- Identification of candidate nasopharyngeal carcinoma serum biomarkers by cancer cell secretome and tissue transcriptome analysis: potential usage of cystatin A for predicting nodal stage and poor prognosis. Proteomics. 2010;10(14):2644-2660.
- [CrossRef] [PubMed] [Google Scholar]
- Cysteine proteinase inhibitors stefin A, stefin B, and cystatin C in sera from patients with colorectal cancer: relation to prognosis. Clin Cancer Res. 2000;6(02):505-511.
- [Google Scholar]
- Proteomic analysis reveals successive aberrations in protein expression from healthy mucosa to invasive head and neck cancer. Oncogene. 2007;26(01):54-64.
- [CrossRef] [PubMed] [Google Scholar]
- Prognostic value of cathepsins B, H, L, D and their endogenous inhibitors stefins A and B in head and neck carcinoma. Biol Chem Hoppe Seyler. 1996;377(06):385-390. F
- [CrossRef] [PubMed] [Google Scholar]
- Discovery of Ca2+-relevant and differentiation-associated genes downregulated in esophageal squamous cell carcinoma using cDNA microarray. Oncogene. 2004;23(06):1291-1299.
- [CrossRef] [PubMed] [Google Scholar]
- Clinicopathological significance of cystatin A expression in progression of esophageal squamous cell carcinoma. Medicine (Baltimore). 2018;97(15):e0357.
- [CrossRef] [PubMed] [Google Scholar]
- Regulation of cisplatin-resistant head and neck squamous cell carcinoma by the SRC/ETS-1 signaling pathway. BMC Cancer. 2019;19(01):485.
- [CrossRef] [PubMed] [Google Scholar]
- Cathepsin B and its inhibitor stefin A in brain tumors. Pflugers Arch. 2000;439(3, Suppl):R122-R123.
- [CrossRef] [Google Scholar]
- Expression of cysteine protease inhibitors stefin A, stefin B, and cystatin C in human lung tumor tissue. Adv Exp Med Biol. 1997;421:259-265.
- [CrossRef] [PubMed] [Google Scholar]
- Cysteine proteinase inhibitor cystatin A in breast cancer. Cancer Res. 1998;58(03):432-436.
- [Google Scholar]
- Lysosomal cathepsins B and L and Stefin A blood levels in patients with hepatocellular carcinoma and/or liver cirrhosis: potential clinical implications. Oncology. 1997;54(01):79-83.
- [CrossRef] [PubMed] [Google Scholar]
- Modulation of cystatin A expression in human airway epithelium related to genotype, smoking, COPD, and lung cancer. Cancer Res. 2011;71(07):2572-2581.
- [CrossRef] [PubMed] [Google Scholar]
- Cystatin A expression reduces bile salt-induced apoptosis in a rat hepatoma cell line. Am J Physiol. 1998;275(04):G723-G730.
- [CrossRef] [PubMed] [Google Scholar]



